Recent Advances in Therapies for Gastrointestinal Stromal Tumour
Gastrointestinal stromal tumour (GIST) is the most common mesenchymal tumour of the gastrointestinal (GI) tract, mainly in the stomach and small intestine. GIST constitutes a distinct group of GI tumours that originate from the interstitial cells of Cajal (ICC), the regulators of peristalsis1. Most of the GISTs occur from the mutations of the gene encoding receptor tyrosine kinases KIT and platelet-derived growth factor receptor £ (PDGFRA). The discovery of these mutations reshapes the biological understanding on the pathogenesis of GIST and, essentially, facilitates the development of therapies in the management of GIST2. While GISTs remain a great therapeutic challenge, potent therapies such as tyrosine kinase inhibitors (TKIs) are emerging to improve the survival and quality of life for patients with GIST.
KIT and PDGFRA - The 2 Switches of GIST
The estimated local prevalence of GISTs was 13.4-15.6 per 100,000 people, with an annual incidence of 1.68-1.96 per 100,000 people, which was comparable to that in the United States3. Former systematic review of population-based studies involving more than 13,000 patients with GIST reported that 55.6% of GISTs were originated in stomach, followed by small bowel (31.8%), colorectal (6.0%), other/various location (5.5%) and oesophagus (0.7%)4. The management of malignant GISTs was once considered as one of the most challenging clinical issues, with very few patients showed response to conventional chemo- and/or radiotherapy. The important findings significantly reshaped the biological understanding of GIST are the recognition of the ICC as the precursor cell and the identification of mutations in KIT and PDGFRA. Mutations in both receptors drive downstream intracellular pathways leading to tumorigenesis2.
KIT is a transmembrane glycoprotein in the receptor tyrosine kinase family that consists of an extracellular domain, a transmembrane domain, a juxtamembrane domain, and a tyrosine kinase domain. The KIT is dimerised and activated upon binding by the stem cell factor (SCF). While normal KIT signalling is required for ICC differentiation and survival, constitutive ligand-independent KIT activation leads to GIST tumorigenesis5. The KIT mutation is seen in approximately 75% of GISTs6. Besides, about 10-15% of GISTs have a gain-of-function mutation in PDGFRA, another receptor tyrosine kinase closely related KIT. Essentially, there is substantial overlap in the signalling pathways downstream of KIT and PDGFRA in GIST (Figure 1)5, suggesting the 2 receptors provide alternative entries into a common tumorigenic pathway. On the other hand, about 10-15% of GISTs those do not have detectable mutations in either KIT or PDGFRA, known as wild-type GIST. Notably, wild-type GISTs constitute about 10% of adult GISTs and up to 85% of paediatric GISTs7. Given the essential role of KIT and PDGFRA in tumorigenesis, targeted therapies aim primarily at one or both of these receptors are developed.
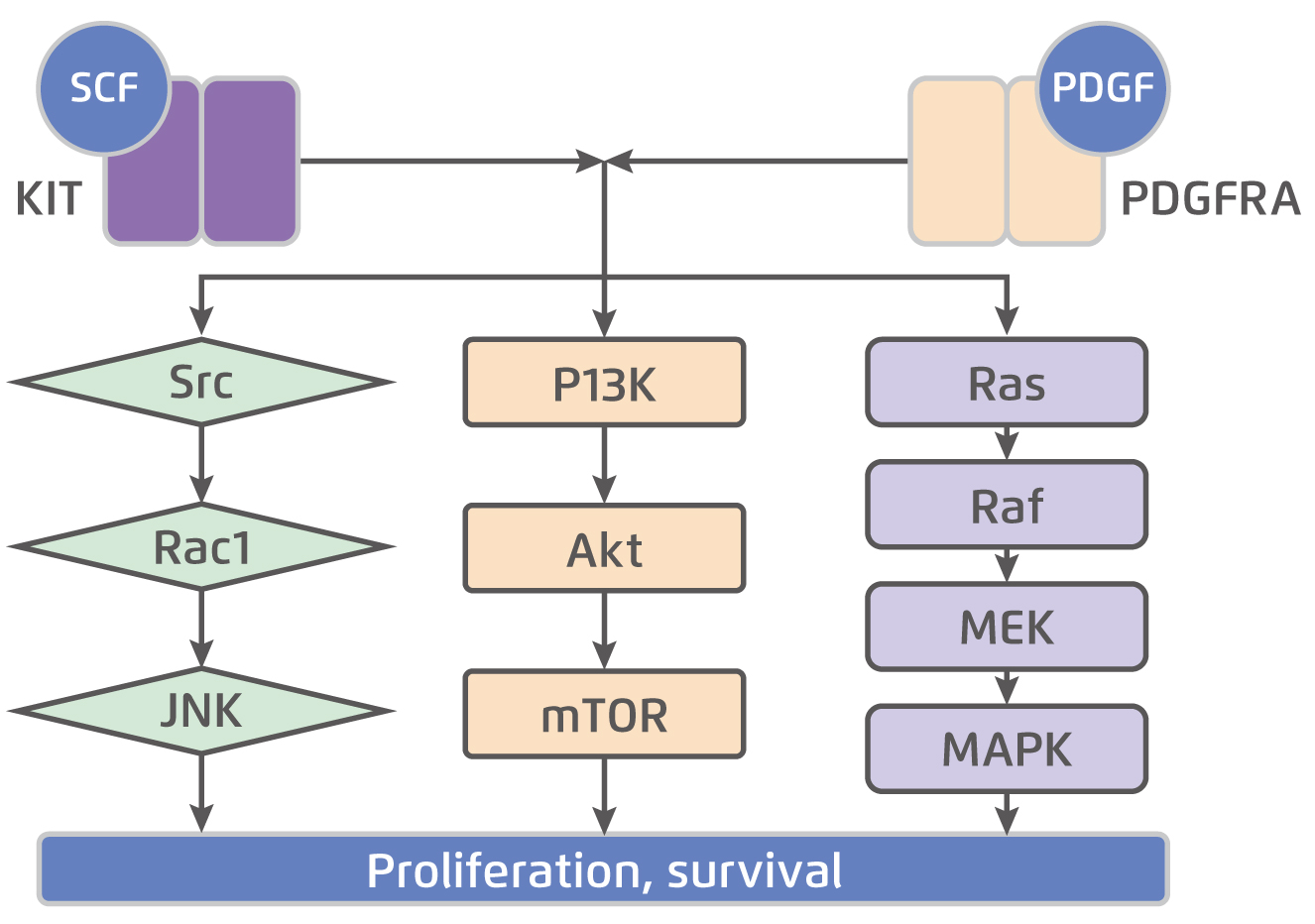
Figure 1.
KIT and PDGFRA downstream signalling pathways5, SCF: stem cell factor; PDGF: platelet-derived growth factor; JNK: c-Jun N-terminal kinase; PI3K: phosphoinositide 3-kinase; mTOR: mammalian target of rapamycin; MEK: mitogen-activated protein kinase; MAPK: mitogen-activated protein kinase
Targeted Therapies for GIST
Although complete tumour resection via surgical approach is warranted for GISTs, radiation or medication therapies would be needed for patients with unresectable GIST. Nonetheless, conventional drug therapies for GIST were reported to be ineffective. For instance, Edmonson et al (2002) demonstrated that a regimen consisted of dacarbazine, mitomycin, doxorubicin, cisplatin, and granulocyte macrophage colony stimulating factor (GM-CSF) yielded objective tumour regression in only one (4.8%) out of 21 patients with GIST, whereas the toxicity was significant with 33% having grade 3 vomiting during treatment. Grade 3 leukopenia occurred in 42%, and grade 3 thrombocytopenia was observed in 68% of the patients8.
The discovery of the roles of KIT and PDGFRA in GIST tumorigenesis triggered the development of therapeutics targeting the markers. The first anti-KIT therapy for GIST was imatinib, a tyrosine kinase inhibitor used initially for chronic myelogenous leukemia that was subsequently found to have both anti-KIT and anti-PDGFRA activity as well9. The therapeutic potential of imatinib was examined in previous clinical trials. In a phase III randomised control trial (RCT) including 746 patients with metastatic or surgically unresectable GIST from 148 centers across the United States and Canada, standard dose of imatinib 400mg daily (n=345) yielded a median progression-free survival (PFS) of 18 months, whereas that yielded by high dose treatment of imatinib, 400mg twice daily (n=349), was 20 months (Figure 2). The median overall survival (OS) achieved by the standard and high dose treatment was 55 and 51 months, respectively (Figure 3)10. The results thus suggested that imatinib was an effective systemic therapy for patients with advanced GIST, but the higher dose did not show any advantage.
Figure 2. PFS achieved by imatinib treatment in advanced GIST10
Figure 3. OS achieved by imatinib treatment in advanced GIST10
DeMatteo et al (2009) further reported the efficacy of imatinib in patients with resectable primary GIST that imatinib 400mg daily (n=359) significantly prolonged recurrence-free survival (RFS) compared with placebo (n=354, 98% vs. 83% at 1 year, hazard ratio [HR]: 0.35, p<0.0001, Figure 4). In addition, adjuvant imatinib was shown to be well-tolerated with a low rate of serious adverse events11. Hence, as compared to placebo, imatinib appears to be a safe and effective treatment in prolonging RFS following the resection of primary GIST.
Figure 4. RFS achieved by imatinib in patients with resectable primary GIST11
Potential Therapeutics Are Emerging
As imatinib inhibits both KIT and PDGFRA signalling, wild-type GISTs without detectable mutations in either gene are generally not responsive to imatinib treatment. Moreover, the majority of patients was reported to develop resistance to treatment with imatinib, mainly due to acquired mutations2. Thus, additional therapies against GIST are investigated. In addition to imatinib, sunitinib, regorafenib and avapritinib have been approved by the FDA for treating GIST.
Sunitinib is a TKI with a broader spectrum of tyrosine kinase inhibition than imatinib. It targets KIT, PDGFRA, VEGFR1 and VEGFR2, FLT3, and RET. Notably, sunitinib is a more potent KIT inhibitor than imatinib12. Regorafenib is another oral multi-targeted TKI with activity on oncogenic pathways (KIT, RET, PDGFRA, FGFR, and BRAF) and angiogenesis pathways (VEGF1-3 and TIE2). Besides, avapritinib is a recently discovered TKI with activity against KIT- or PDGFRA-resistant mutations6. The approval of these TKIs was based on the improved outcomes demonstrated in phase III RCTs. Moreover, there are other TKIs being studied in patients resistant to imatinib, sunitinib and regorafenib. For instance, ripretinib is a TKI inhibiting phosphorylation of KIT in all tested mutants that were resistant to imatinib and sunitinib. It was reported to be more potent than regorafenib in inhibiting KIT mutants as well13. In the phase III INVICTUS trial, ripretinib (n=85, median PFS: 6.3 months) significantly prolonged median PFS compared with placebo (n=44, median PFS: 1.0 month, HR: 0.15, p<0.0001) in patients with advanced GIST with disease progress or intolerance to imatinib, sunitinib, and/or regorafenib. The results also revealed the acceptable safety profile of ripretinib14. Thus, ripretinib has recently been approved by the FDA for treating advanced GIST15.
Besides TKIs, the role of immunotherapy in the management of GIST has been studied extensively. Recent case report suggested that treatment with nivolumab, an anti-programmed cell death protein 1 (PD-1) antibody, yielded durable disease control of more than 30 months in a patient with wild-type GIST, whose disease was not controlled by TKI treatments with imatinib, sunitinib, regorafenib and sorafenib16. Moreover, a recent phase II trial involving 27 patients with advanced/metaststic GIST refractory to at least imatinib demonstrated the efficacies of nivolumab monotherapy and nivolumab combined with ipilimumab in controlling GIST. The results showed that 1 patient out of 12 (8.3%) had a partial response and 2 out of 12 (16.7%) had stable disease (SD) in the combination arm, while 7 SD were seen among 15 patients (46.7%) treated with nivolumab. The median PFS in the nivolumab and in combination arm were 8.57 and 9.10 weeks, respectively17.
Clinically, management of GIST remains challenging. Nonetheless, the approval of new potent TKIs offers more treatment options for controlling the disease. As mentioned, other potential TKIs are emerging to add therapeutic values in treating certain resistant GISTs. In prospective, further trials evaluating therapeutic potentials of agents including immunotherapy, either as monotherapy or in combination treatments, would likely help managing specific subsets of GIST patients.
References:
1. Logroño et al. Cancer Biol. Ther. 2004; 3: 251-8. 2. Mei et al. Front. Oncol. 2018; 8. 3. Chan et al. World J Gastroenterol 2006; 12: 2223-8. 4. Søreide et al. Cancer Epidemiol. 2016; 40: 39-46. 5. Li et al. Onco Targets Ther 2019; 12: 5123-33. 6. Arshad et al. Expert Rev. Anticancer Ther. 2020; 20: 279-88. 7. Pappo et al. Hematol. Oncol. Clin. North Am. 2009; 23: 15-34. 8. Edmonson et al. Cancer Invest 2002; 20: 605-12. 9. Tuveson et al. Oncogene 2001; 20: 5054-8. 10. Blanke et al. J Clin Oncol 2008; 26: 626-32. 11. DeMatteo et al. Lancet 2009; 373: 1097-104. 12. Marrari et al. Arch Pathol Lab Med 2012; 136: 483-9. 13. Martin-Broto et al. Curr Opin Oncol 2020; 32: 314-20. 14. Blay et al. Lancet Oncol 2020; 21: 923-34. 15. FDA. Novel Drug Approvals for 2020. 2020. 16. Schroeder et al. Oncoimmunology 2020; 9. 17. Singh et al. J Clin Oncol 2019; 37: 11017-11017.