Regulation of the JAK/STAT Pathway by SOCS Proteins in Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic, polyarticular, chronic, progressive inflammatory musculoskeletal disorder of the synovial joint. Essentially, RA-associated complications can occur in various organs including the heart, kidney, eye and etc. Clinically, RA is characterised by abnormal innate, cellular and humoral immunity as reflected by the abnormal proliferation kinetics of immune cells such as T-lymphocytes, marcophages and accessory-antigen presenting cells1. Suppressor of cytokine signaling (SOCS) proteins are mainly induced by various cytokines and recent evidence suggests that SOCS proteins are often dysregulated in a wide variety of autoimmune diseases including RA2. In this article, the role of SOCS proteins in the regulation of cytokine signaling and its implication on the development of therapeutics for RA are highlighted.
The JAK/STAT Signaling Cascade
Janus-associated kinases (JAK) are cytoplasmic tyrosine kinases mediating a variety of cytokine signals, which further affect cellular growth, differentiation, survival and death, predominantly in haematopoiesis and immune response. Dysregulated JAK activity is involved in haematological malignancies, autoimmune disorders and immunodeficient conditions and has been implicated in the pathogenesis of a subset of solid tumours3.
Currently, there are 4 members identified in the JAK family, namely JAK1, JAK2, JAK3 and TYK2. The JAK proteins are associated with dimeric cytokine receptors and are activated upon ligand binding leading to JAK autophosphorylation followed by phosphorylation of intracellular receptor tyrosines creating binding sites for SH2 domain-containing proteins such as signal transducers and activators of transcription (STAT) proteins. The activated STAT proteins dimerise and translocate into the cell nucleus to influence DNA transcription, thus regulating gene expression (Figure 1)4. The various combinations of JAK pairs recruit different STAT proteins, of which there are up to 6 types, and this allows for the wide range of downstream activities in the JAK-STAT pathways5.

Figure 1. The JAK/STAT signaling and inhibitors (circled)4
RA Therapies Targeting JAK/STAT Pathway
In RA synovial joints, the normal synovium becomes hyperplastic as a result of the interactions between immune and non-immune cells under the direction of elevated levels of various chemokines and adhesion proteins. Additionally, the levels of proinflammatory cytokines and growth factors are significantly increased. Eventually, the destruction of articular cartilage and erosion of subchrondral bone occur resulting in synovial joint failure6,7.
The attention on the role played by JAK/STAT pathway in RA has been initiated by the fact that several cytokines implicated in RA pathogenesis, such as IL-6, IL-15 and interferons (IFNs), activate the JAK/STAT pathway, while STAT3 is constitutively activated in RA8. Hence, selective inhibition of JAKs has been identified as an important strategy for the treatment of autoimmune disorders. Of note, blockade of JAK/STAT pathway by small molecule inhibitor (SMI), which inhibits phosphorylation of JAK1 and JAK3, subsequent STAT1 as well as STAT1-inducible genes, contributes to the efficient propagation of anti-inflammatory effects for the treatment of RA9. Essentially, tofacitinib, a JAK3-selective SMI, was approved by the US FDA for the medical therapy of moderate-to-severe active RA in patients whose response to methotrexate was deemed to be inadequate1. The approval of tofacitinib has encouraged further investigations on molecular mechanisms of RA progression and their regulation through the JAK/STAT pathway, hence the development of JAK-selective SMIs.
The SOCS Proteins
The SOCS family consists of 8 proteins, SOCS1-SOCS7 and cytokine-inducible SH2-domain (CIS), each of which has a central SH2 domain, an amino-terminal domain of varying length and sequence and a carboxy-terminal 40-amino-acid module known as the SOCS box (Figure 2)10. It has been reported that the expression levels of SOCS are low in the basal state, but are rapidly induced by the JAK/STAT pathway. The upregulated SOCS protein expression in turn triggers a negative feedback process for over-activated cytokine signaling via 3 primary mechanisms. Firstly, SOCS proteins can inhibit JAK activity directly by binding to either the signaling receptor or to the JAK activation loop11. Besides, SOCS proteins suppress cytokine signaling through substrate competition. For instance, CIS would compete with tyrosine residues of cytokine receptors which provide docking sites for STAT512. Moreover, SOCS proteins suppress signaling pathways by targeting associated substrates for degradation through ubiquitination (Figure 3)2. Thus, SOCS proteins are involved in the negative regulation of the JAK/STAT pathway. Importantly, it has been reported that SOCS proteins would regulate other signaling pathways such as phosphoinositide 3-kinase (PI3K)-AKT and nuclear factor-κB (NF-κB) signaling as well.

Figure 2. The structure of SOCS protein family members10
PEST: proline, glutamic acid, serine and threonine; ESS: extended SH2 subdomain; KIR: kinase inhibitory region
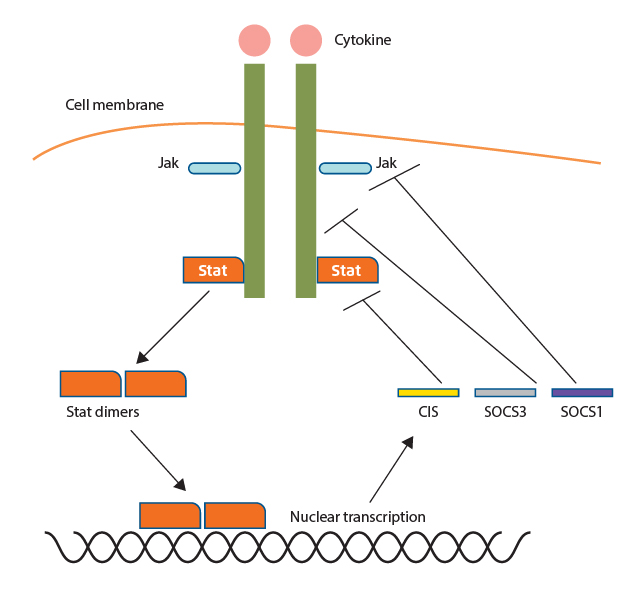
Figure 3. Negative cytokine signaling regulation by SOCS proteins2
Therapeutic Implications of SOCS Proteins
The cell-intrinsic role of SOCS proteins in the regulation of cytokine signaling indicates that they can be targeted for the treatment of autoimmune diseases which are associated with uncontrolled cytokine signaling. For instance, the upregulation of SOCS proteins would be a treatment strategy for suppression of detrimental prolonged cytokine signaling. Veenbergen et al (2011) demonstrated in a mice model that intravenous treatment with adenoviruses encoding SOCS3 before onset of collagen-induced arthritis (CIA) would effectively impair the development of the disease via controlling the immunostimulatory capacities of antigen-presenting cells (APCs), such as invariant natural killer T (iNKT) cells13.
Besides negative feedback of cytokine signaling, SOCS proteins can act as antagonists of cytokines as well. Waiboci et al (2007) synthesised p-JAK2, a phosphorylated human JAK2 peptide which had SOCS1 antagonistic properties, as it could suppress SOCS1-mediated inhibition of STAT3 activation. Of note, p-JAK2 was reported to enhance suboptimal IFN-γ-induced antiviral activity14. Hence, the findings provide insight on the clinical applications of therapeutics which mimic the functions and/or modulate the expressions of SOCS proteins.
The regulation of cell growth and differentiation is highly complicated whereas the JAK/STAT signaling cascade plays an important role in mediating a wide spectrum of cellular functions. However, uncontrolled JAK/STAT activities are expected to be responsible for various autoimmune and inflammatory disorders including RA. Although much remains to be explored regarding the function of SOCS proteins in RA, previous investigations demonstrated the therapeutic potential of SOCS proteins and their analogues in controlling RA through regulating the JAK/STAT pathway.
References
1. Malemud. Ther Adv Musculoskelet Dis. 2018;10(5-6):117-127. 2. Liang et al. Eur J Immunol. 2014;44(5):1265-1275. 3. Meyer et al. Clin Cancer Res. 2014;20(8):2051-2059. 4. Aittomäki et al. Basic Clin Pharmacol Toxicol. 2014;114(1):18-23. 5. Hsu et al. J Immunol Res. 2014;2014:283617. 6. Brennan et al. Curr Opin Rheumatol. 2007;19(3):296-301. 7. Imboden. Annu Rev Pathol Mech Dis. 2009;4(1):417-434. 8. Ivashkiv and Hu. Arthritis Rheum. 2003;48(8):2092-2096. 9. Tanaka. J Biochem. 2015;158(3):173-179. 10. Kubo et al. Nat Immunol. 2003;4(12):1169-1176. 11. Yasukawa et al EMBO J. 1999;18(5):1309-1320. 12. Yang et al. Nat Immunol. 2013;14(7):732-740. 13. Veenbergen et al. Ann Rheum Dis. 2011;70(12):2167-2175. 14. Waiboci et al. J Immunol. 2007;178(8):5058-5068.