Recent Advancement in Pharmacological Control of Infantile Onset Pompe Disease
Pompe disease (PD), is a rare severe metabolic disorder characterised by a deficiency of acid α-glucosidase (GAA) and lysosomal accumulation of glycogen, particularly in cardiac and skeletal muscle, leading to cellular dysfunction and muscle damage1. The incidence of PD is estimated to be 1 in 40,000 live births, whereas higher incidences have been reported in Austria (1 in 8,684) and Taiwan (1 in 17,000)2. Essentially, for the infantile onset form of PD (IOPD), rapid progression of disease leading to death within the first year of life would be resulted without treatment1. Currently, enzyme replacement therapy (ERT) with recombinant human GAA is the only treatment available for PD and the treatment significantly improves the outcomes of patients. Thanks to the better understanding on the pathophysiology of PD, a new generation of recombinant GAA, avalglucosidase alfa (neoGAA), has recently been developed and is expected to further optimise the management of PD.
IOPD at a Glance
Pompe disease (PD), also known as glycogen storage disease type II or acid maltase deficiency, is caused by biallelic mutations in the gene located on chromosome 17q25 encoding for the lysosomal hydrolase GAA. There are two classic forms of PD identified in the literature, namely infantile- (IOPD) and late onset PD (LOPD). LOPD can be further differentiated into childhood, juvenile, and adult-onset forms1.
While the severity of PD depends on the amount of residual GAA activity, IOPD is characterised by virtually complete absence of GAA activity. The absence of GAA activity leads to rapidly progressive muscle weakness, hypotonia, macroglossia, hepatomegaly, and progressive hypertrophic cardiomyopathy (HCM, Figure 1)3. In addition, feeding difficulties and respiratory tract infections have been reported in patients with IOPD as well4. Essentially, the symptoms of IOPD typically present during the first few weeks of life, whereas death usually within the first 12 months of life if untreated5.
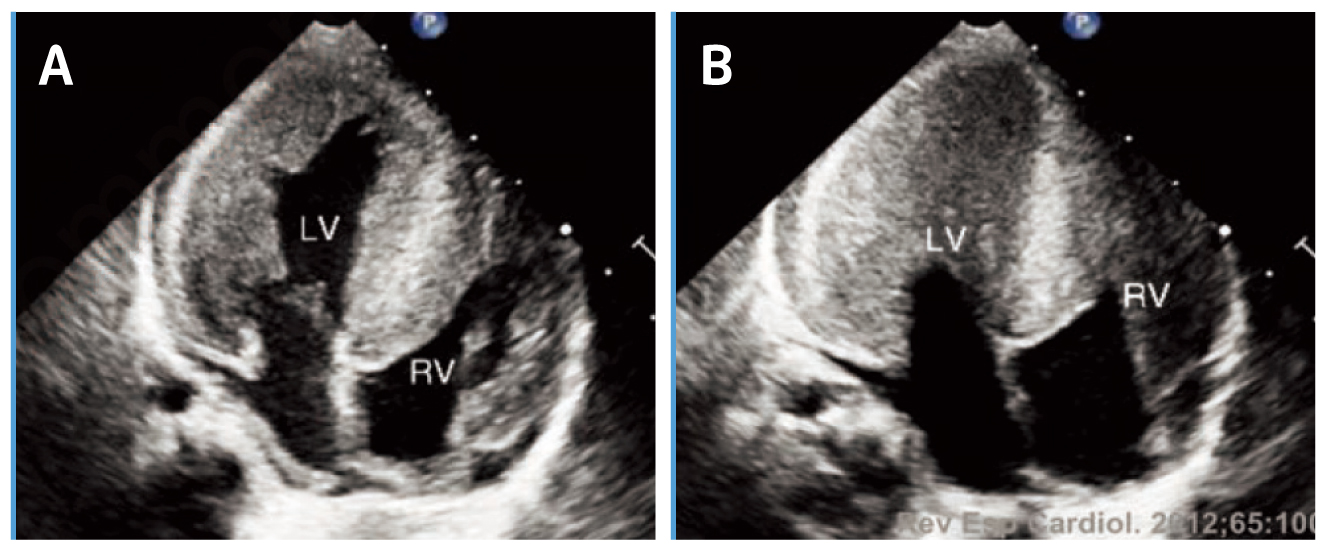
Figure 1. HCM in a patient with IOPD during (A) diastole and (B) systole6, LV=left ventricle, RV=right ventricle
ERT in IOPD
ERT with recombinant human GAA (rhGAA, alglucosidase alpha) is the only treatment currently available for PD. The treatment has been reported to reverse the cardiomyopathy developed, improve clinical course and the expected outcome of patients with either IOPD7,8 or LOPD9. For instance, a clinical trial by Chien et al (2015) involving 10 patients with classic IOPD demonstrated that, after a median treatment time of 63 months, all rhGAA-treated patients could walk independently, and none required mechanical ventilation. Although muscle weakness over pelvic girdle appeared gradually after 2 years of age, all patients had motor capability sufficient for participating in daily activities (Figure 2). Ptosis was present in one-half of the patients, and speech disorders were common. Nonetheless, no significant anti-rhGAA antibody interference was developed and the treatment was shown to be safe and well tolerated8. The results thus showed the significant improvement in prognosis in the patients with IOPD.
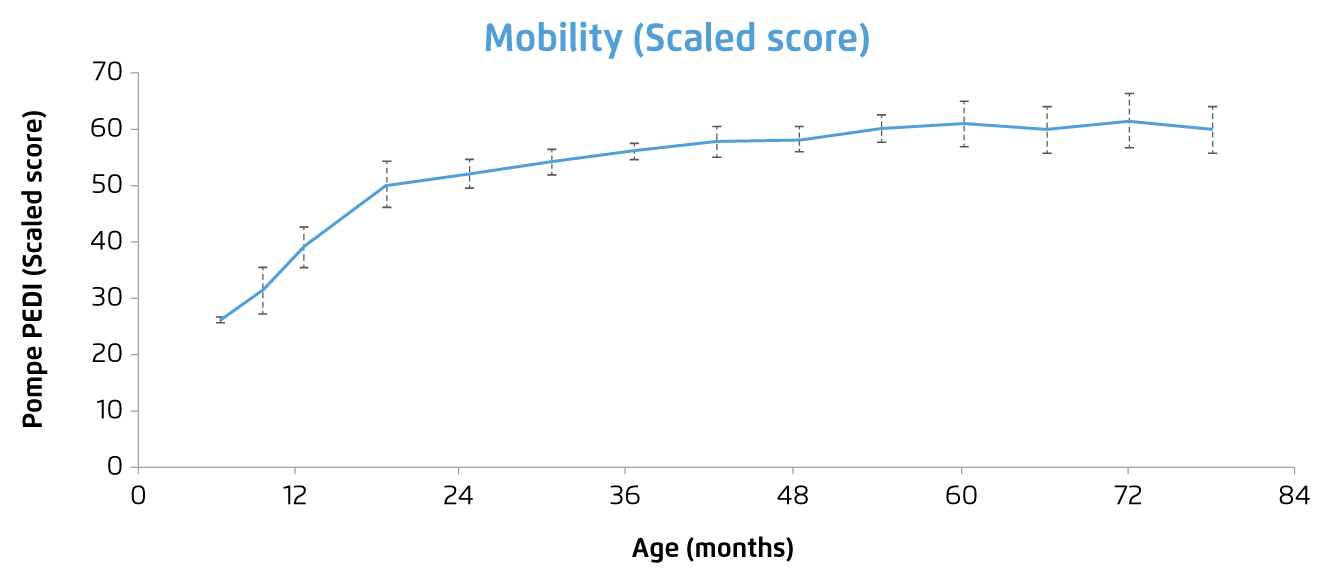
Figure 2. Change in motor function score of IOPD patients received ERT8, note: greater scores reflect superior capability
As demonstrated in the results above, ERT may not resolve the symptoms of IOPD completely. Former report suggested that optimal outcomes would be hampered in infants started on ERT after five months of age, or in those with a marked increase in the left ventricular mass index10. This highlights the importance of early diagnosis and prompt treatment for IOPD patients.
Upon the diagnosis of IOPD is documented, determination of patients’ cross-reactive immunologic material (CRIM) status is crucial before initiation of ERT. Patients with residual GAA protein are classified as CRIM-positive while those who completely lack the enzyme are classified as CRIM-negative11. Of importance, CRIM-negative individuals may develop higher anti-rhGAA antibodies during ERT and immunomodulation therapy may be required, ideally prior to the first infusion6.
The New Generation of Recombinant GAA for PD
Recent investigations on physiological responses to rhGAA treatment in PD patients lead to a better understanding on the pathophysiology of the disease and hence facilitate further development on new therapeutics. Notably, alglucosidase alpha has been reported to clear glycogen storage in cardiac muscle more effectively than in skeletal muscle, which partially reflects the tissue differences in cation-independent mannose 6-phosphate (CIM6P)/insulin growth factor II (IGFII) receptor expression and enzyme uptake12. The cell-surface CIM6P/IGFII receptor mediates cellular uptake of exogenous GAA and targets it to the lysosomal compartment13. Thus, increasing M6P on the recombinant enzyme may increase achieved skeletal muscle uptake14. These findings led to the development of neoGAA, a second-generation, glycoengineered recombinant GAA with increased bis-M6P levels on the molecule.
In the Phase III COMET trial, neoGAA was demonstrated to achieve greater improvements in pulmonary function and 6-minute walk test results in patients with LOPD as compared to alglucosidase alpha. Moreover, neoGAA exhibited a more favourable safety profile as well15. Besides, clinical trials investigating the efficacy and safety of neoGAA in IOPD are emerging. Of note, the safety and immunogenicity of neoGAA in IOPD patients with clinical decline on alglucosidase alpha have been evaluated in the Phase II mini-COMET trial, whereas the interim results demonstrated that neoGAA was well-tolerated in the ERT-experienced IOPD patients16. Yet, further clinical data confirming the long-term safety and efficacy of neoGAA in IOPD are needed. Nonetheless, ERT with neoGAA for symptomatic IOPD patients at least 1 year old and have received alglucosidase alpha for at least 6 months has been approved in the United Kingdom by virtue of the demonstrated improvement in IOPD patients as well as the limited treatment option17.
Newborn Screening - The Key for Optimal Outcomes
As mentioned, early initiation of ERT in IOPD markedly improves survival, reduces the need for ventilation, results in earlier independent walking, and enhances quality of life7. Nonetheless, timely diagnosis of PD and subsequent treatment are hampered in reality because of the lack of screening tests and limited awareness on the rare disease amongst healthcare professionals18. This highlights the importance of newborn screening (NBS) for rare disease including PD. A recent report on the outcomes of NBS for PD in Taiwan suggested that Pompe NBS with dried blood spot (DBS) test was able to identify patients with IOPD as well as underdiagnosed LOPD or even asymptomatic GAA-deficient individuals19.
In summary, former clinical data have demonstrated the efficacy and promising safety profile of ERT, whereas effective and safe therapies for patients with PD are emerging. Nonetheless, early and accurate diagnosis is the prerequisite for timely treatment, which is still a major clinical challenge in the management of PD. While many children with PD are not diagnosed until after referral to a clinical specialist, it is sad but true that many of the children do not survive the time it takes to confirm the diagnosis. Hence, NBS for rare inherited diseases including PD is worth considering.
References
1. Kishnani et al. Genet. Med.2006; 8: 267-88. 2. Zhao et al. Orphanet J Rare Dis 2019 141 2019; 14: 1-7. 3. Martínez et al. Medicine (United States) 2017; 96(51): e9186. 4. Chu et al. Neuromuscul Disord 2016; 26: 873-9. 5. Hahn and Schänzer. Ann Transl Med 2019; 7: 283-283. 6. Felice . Cardiogenetics 2017; 7: 6857. 7. Chien et al. Pediatrics 2009; 124: e1116-25. 8. Chien et al. J Pediatr 2015; 166(4): 985-91.e1-2. 9. Milverton et al. J Inherit Metab Dis 2019; 42: 57-65. 10. Suzuki et al. Clin Genet 1988; 33: 376-85. 11. Kronn et al. Pediatrics 2017; 140: S24-45. 12. Pena et al. Neuromuscul Disord 2019; 29: 167-86. 13. Koeberl et al.Mol Genet Metab 2011; 103: 107. 14. McVie-Wylie et al. Mol Genet Metab 2008; 94: 448. 15. Kushlaf et al.Neurology 2021; 96. 16. Chien et al. J Inherit Metab Dis 2019; 42: 229. 17. Medicines and Healthcare products Regulatory Agency. Early Access to Medicines Scientific Opinion-Public Assessment Report. 2021. 18. Molster et al. Orphanet J Rare Dis 2016; 11: 30. 19. Sawada et al. Int J Neonatal Screen 2020; 6(2): 31.