The treatment of hemophilia has evolved dramatically over the past century, transitioning from rudimentary blood transfusions to sophisticated biologics and gene therapies. Blood transfusions are blood clotting products that replace the missing or deficient clotting proteins. Biologics, such as the factor mimetic emicizumab, work by replacing the function of factor VIII, offering less frequent, subcutaneous treatment options for hemophilia A; while gene therapy uses viral vectors to deliver the correct gene into liver cells, enabling them to make clotting factor IX or VIII.1 Here is a brief overview of this evolution, culminating in the recent approval of marstacimab, a rebalancing agent, as a novel non-factor therapy.
In 1828, German physician Dr. Johann Lukas Schönlein and his student Friedrich Hopff coined the term "haemorrhaphilia" to describe the bleeding disorder, which was later shortened to "haemophilia" (British English) or "hemophilia" (American English). This term, derived from Greek words for "blood" and "love of" (haîma and philía), refers to the inherited genetic condition where the body's blood does not clot properly, leading to excessive bleeding.2
By the late 1950s and early 1960s, fresh frozen plasma was transfused in patients in the hospital. However, each bag of the plasma contained so little of the necessary clotting factor that huge volumes of it had to be administered. Many children experienced severe joint bleeds that were crippling. Intracranial hemorrhage could be fatal. By 1960, the life expectancy for a person with severe hemophilia was less than 20 years old.2
Not until the 1970s did freeze-dried powdered concentrates containing factor VIII and IX become available. Factor concentrates revolutionized hemophilia care because they could be stored at home, allowing patients to “self-infuse” factor products, alleviating trips to the hospital for treatment.2 However, plasma-derived coagulation factor concentrates, prepared using traditional manufacturing processes, were found to transmit viral diseases, especially AIDS, hepatitis B and hepatitis C to patients.3 Hemophilia patients in China were among the first in the country to be infected with HIV, contracting the virus in the mid-1980s from contaminated US-imported factor VIII concentrates. This led to a significant HIV epidemic within the hemophilia community in China; and the overwhelming impact was felt into the next few decades.4
Treatment for hemophilia advanced in the 1990s. Factor products became safer as tighter screening methods were implemented and advanced methods of viral inactivation were used.2 Additionally, in 1992, the US Food and Drug Administration (FDA) approved the recombinant factor VIII concentrate, which is genetically engineered using DNA technology. Recombinant factor concentrates do not contain any plasma or albumin, and therefore, cannot spread any bloodborne viruses.1 However, some patients develop inhibitors, or antibodies, to infused factor products. Inhibitors make it more difficult to stop a bleeding episode because they prevent the treatment from working. According to a 2021 meta-analysis, recombinant factor VIII is associated with a higher risk of overall and high-responding inhibitor development compared to plasma-derived FVIII in previously untreated patients with hemophilia A.5 This calls for the development of a bypassing agent which may offer these patients an alternative option to help stop bleeds and joint damage.
In 2017, approval of emicizumab, a bispecific monoclonal antibody mimicking factor VIII, revolutionized prophylaxis for hemophilia A. This agent is indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A with factor VIII inhibitors.6
In hemophilia, the unmet needs regarding adherence to prophylaxis and lack of effective long-term prophylaxis regimens, especially in patients with inhibitors, led to the production of emicizumab, the first non-factor medicine.7 Emicizumab restores the function of missing activated factor VIII (FVIIIa) by bridging activated factor IX and factor X, mimicking FVIIIa independently of factor VIII levels. Consequently, emicizumab promotes effective hemostasis in patients with hemophilia A.8
The promise of emicizumab as a convenient-to-administer, subcutaneous, non-factor FVIIIa mimetic with durable effectiveness was demonstrated in HAVEN 1 and 2 clinical trials.9,10 The most concerning of the adverse events reported with emicizumab during clinical trials has been the occurrence of thromboembolic events and thrombotic microangiopathy. Critically, these complications were associated with concomitant replacement therapy with activated prothrombin complex concentrate (aPCC) at cumulative doses over 100 U/kg/day for more than 1 day.7 Furthermore, emicizumab was also associated with the development of antidrug antibodies in 5.1% of patients (34/668) who participated in the HAVEN 1-4 clinical trials, which were presumed to be responsible for decreasing the plasma concentration of emicizumab and its subsequent loss of efficacy.11
As of 2022, 62 trials on gene therapy for hemophilia were underway.12 On 29 June 2023, the FDA approved roctavian, the first gene therapy for adults with severe hemophilia A.13 Gene therapy for hemophilia is based on the transfer of a non-pathogenic and non-replicating recombinant adeno-associated virus (AAV), the viral DNA of which has been replaced by a bioengineered gene cassette, with a tissue-specific promoter and other regulatory elements. Following intravenous infusion and subsequent cell transduction, endocytosis and import into the nucleus occurs, where the genetic material is released as episomal DNA. The therapeutic gene can then be expressed, thereby resulting in the production of the therapeutic protein, such as factor VIII or IX.12
Data from non-randomized phase 1 to phase 3 trials reveal an adequate expression of factors VIII and IX in patients with mostly severe hemophilia A or B. Even though they were no longer receiving prophylactic treatment, most patients experienced a considerable reduction, by 53% to 96%, in the number of bleedings compared to previous therapy. Persistently elevated factor levels have been described for up to six years in hemophilia A and up to eight years in hemophilia B.14,15,16
Disadvantages of AAV include pre-existing neutralizing antibodies to AAV, possible liver reactions, and a presumably non-permanent response; furthermore, due to the generation of neutralizing antibodies, the treatment can only be performed once. In contrast to other viral vectors, such as lentiviruses, AVV are largely not integrated in an individual’s genetic information, explaining why hemophilia can still be passed on despite gene therapy.12
On 11 October 2024, marstacimab was approved by FDA for patients ≥12 years with hemophilia A or B without inhibitors.17 Although replacement treatment appears to be effective for hemophilia, the frequent venous infusions required for prophylaxis are burdensome to patients. Even with treatment, some patients still experience long-term joint damage from bleeds.18
The first non-factor prophylactic agent approved for hemophilia was emicizumab. It mimics the activity of factor VIII, so it is only indicated for hemophilia A and not hemophilia B. In contrast, the monoclonal antibody marstacimab inhibits the activity of tissue factor pathway inhibitor (TFPI), a naturally occurring anticoagulation protein. As kind of a global hemostatic product, marstacimab increases the capacity to clot, independent of the level of any factor, rebalancing the hemophilia teeter-totter.18
In BASIS, a phase III, open-label, crossover trial, marstacimab was studied as a prophylactic treatment for severe hemophilia A and moderately severe to severe hemophilia B. In one cohort of the study, 116 patients without inhibitor antibodies were treated with weekly subcutaneous marstacimab in the active treatment phase of the study.19 In the patients who had previously received on-demand clotting factors, the mean annualized bleeding rate (ABR) decreased from 39.9% to 3.2% with marstacimab treatment, corresponding to a reduction of 92%. In the group that had previously been receiving routine prophylaxis with replacement factors, the ABR decreased from 7.9% to 5.1%, corresponding to a decrease of 35.5% (Figure 1).19 The BASIS trial also included patients with inhibitors to coagulation factors. These data have not been fully reported, but in June positive findings in this setting were also announced.18
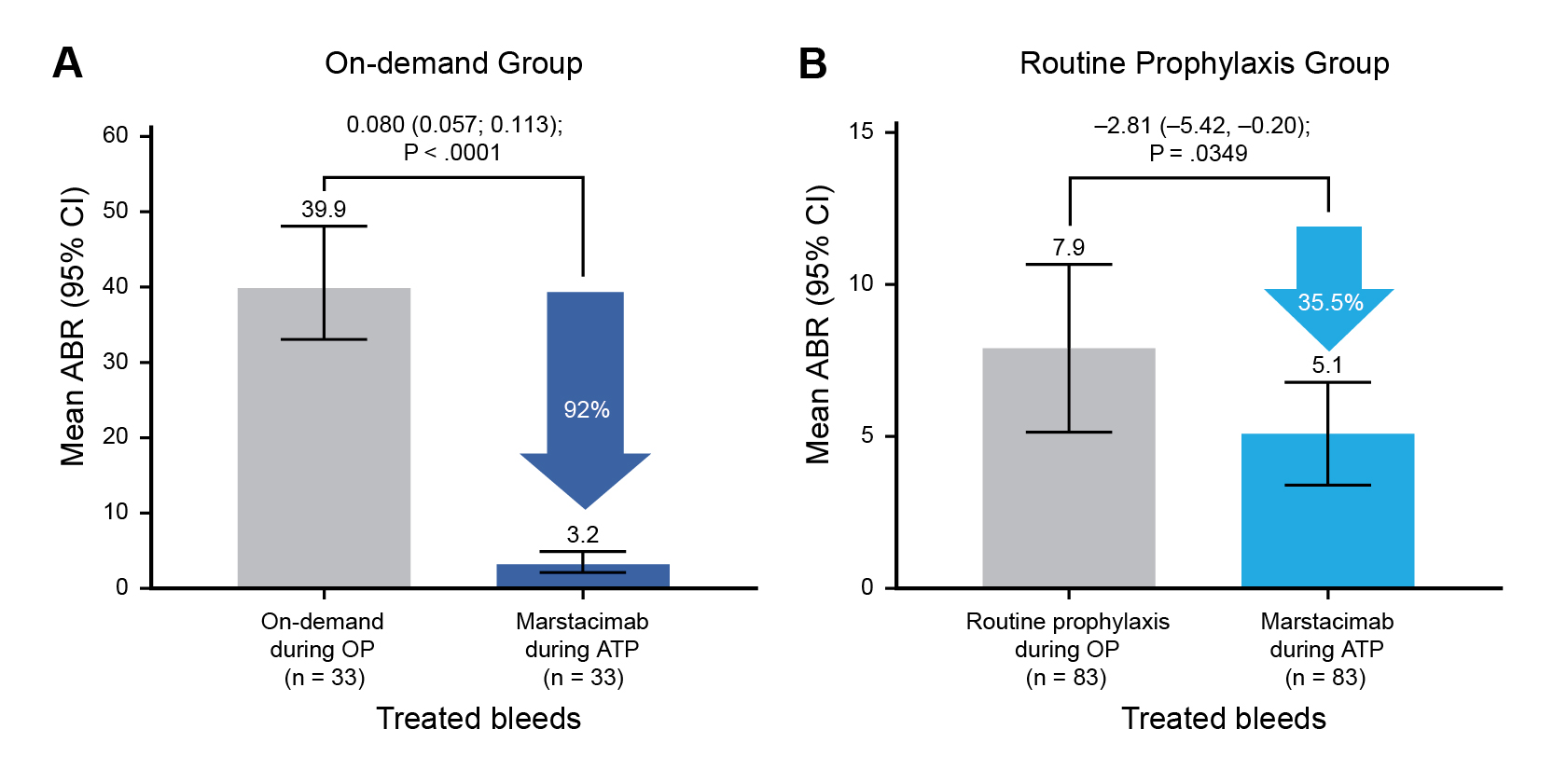
ATP=active treatment phase; OP=observational phase
Figure 1. Marstacimab prophylaxis reduced bleeding events compared with prior factor replacement therapy19
Administered subcutaneously at a weekly fixed dose regardless of weight or age, marstacimab supports consistent thrombin generation and bleeding prevention in hemophilia A and B patients.20 Although clearly an important innovation, especially for hemophilia B, more long-term data regarding efficacy and safety are needed to see how the therapy will fit with other treatment options.
The journey of hemophilia treatment reflects a remarkable trajectory from rudimentary transfusions to cutting-edge biologics and gene therapies. Early reliance on plasma products, though life-saving, carried significant risks. The advent of recombinant factor therapies in the 1990s marked a turning point in safety and efficacy, followed by innovations like factor mimetic therapies such as emicizumab. The recent approval of marstacimab represents a new frontier in hemophilia care. As a subcutaneous, non-factor therapy targeting TFPI, it offers a convenient and effective option for both hemophilia A and B patients without inhibitors. Its fixed dosing and weekly administration simplify treatment, potentially improving adherence and quality of life. Looking ahead, marstacimab complements a growing arsenal of therapies that aim not just to manage bleeding but to transform hemophilia into a condition with minimal daily burden. With gene therapy on the horizon and global access improving, the future of hemophilia care is more hopeful than ever.
References
1. CDC. Treatment of Hemophilia. 13 November 2024. Available from: https://www.cdc.gov/hemophilia/treatment/index.html. [Accessed 29 September 2025]. 2. National Bleeding Disorders Foundation. History: From 2 AD to the present. Available from: https://www.bleeding.org/bleeding-disorders-a-z/overview/history. [Accessed 29 September 2025]. 3. Burnouf T. Biologicals. 1992;20(2):91–100. 4. Wu Z, et al. Curr HIV/AIDS Rep. 2019;16:458–66. 5. Kohar K, et al. Cureus. 2022;14(6):e26015. 6. FDA. FDA approves emicizumab-kxwh for prevention and reduction of bleeding in patients with hemophilia A with factor VIII inhibitors. 16 November 2017. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-emicizumab-kxwh-prevention-and-reduction-bleeding-patients-hemophilia-factor-viii. [Accessed 29 September 2025]. 7. Andrade PEA, et al. Haematologica. 2024;109(5):13344–7. 8. Parisi K and Kumar A. Emicizumab. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559180/. [Accessed 29 September 2025]. 9. Oldenburg J, et al. N Engl J Med. 2017;377(9):809–18. 10. Young G, et al. Blood. 2019;134(24):2127–38. 11. Callaghan MU, et al. Blood. 2021;137(16):2231–42. 12. Miesbach W, et al. Dtsch Arztebl Int. 2022;119(51-52):887–94. 13. FDA. DA Approves First Gene Therapy for Adults with Severe Hemophilia A. 29 June 2023. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-adults-severe-hemophilia. [Accessed 29 September 2025]. 14. George LA, et al. N Engl J Med. 2021;385:1961–73. 15. Nathwani AC, et al. N Engl J Med. 2011;365:2357–65. 16. Miesbach W, et al. Blood. 2018;131:1022–31. 17. FDA. FDA Approves New Treatment for Hemophilia A or B. 11 October 2024. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-hemophilia-or-b. [Accessed 29 September 2025]. 18. Hickman RJ. Marstacimab Provides New Prophylactic Option for Hemophilia A, B. September 2025. Available from: https://ashpublications.org/ashclinicalnews/news/8726/Marstacimab-Provides-New-Prophylactic-Option-for. [Accessed 29 September 2025]. 19. Matino D, et al. Blood. 2025;146(14):1654–63. 20. Mahlangu J. Biologics: Targets and Therapy. 2025;19 379–86.