Atherosclerotic cardiovascular disease (ASCVD) is one of the main causes of death globally, accounting for 31% of global deaths attributed by ASCVD. Furthermore, cardiovascular disease (CVD) accounts for 1 in every 5 deaths, globally1. The most common risk factors for ASCVD include hypercholesterolaemia, hypertension, diabetes mellitus, cigarette smoking, age (males older above the age of 45 and females above the age of 55)2. Traditionally, it is believed that among all the modifiable risk factors for the CVD, serum levels of cholesterol, particularly the low-density lipoprotein cholesterol (LDL-C) is associated with highest attributable risk for the occurrence of ischaemic heart disease (IHD)3. In fact, clinical trials have shown a reduction of 1.0mmol/L in LDL-C equated to a 20% risk reduction in major vascular events4. Aggressive LDL-C reduction is often advocated with first- and second-line treatments for primary and secondary prevention initiated early5. Nonetheless, despite significant emphasis given to LDL-C reduction, recent epidemiological studies have shown LDL-C may not be the only mediator in the development of CVD6.
In fact, a lesser known lipoprotein (a) [Lp(a)], has recently shown to promote ASCVD, with very high levels of serum Lp(a) (>190 mg/dL or >430 nmol/L) being associated with an approximately 2.5-fold increase in ASCVD risk compared to normal Lp(a) levels (<14 mg/dL)7,8. So, what is Lp(a) and what is its role in CVD? Lp(a) is related to LDL-C as both share similar size, lipid content, and the presence of apolipoprotein (apop) B100. Contrary to LDL-C, Lp(a) contains one apo(a) of variable length that is linked by a single disulfide bond to apo B1009. Moreover, the apo(a) has a similar physical structure to that of tissue plasminogen activator (TPA) and plasminogen; therefore, it is able to actively complete with plasminogen for specific binding sites, thereby, leading to a reduction in the fibrinolysis process7. According to the European Atherosclerosis Society (EAS), approximately 20% to 25% of the global population have an Lp(a) level of 50 mg/dL or greater, which confers to an increased CV risk despite traditional risk factor optimisation10. Furthermore, statins has shown to increase the Lp(a) levels by 10-20%, which may further aggravate the CVD risk5.
Lp(a) was first identified by Dr Kare Berg in 1963 and is genetically inherited. Epidemiologic and observational studies recently conducted suggest a potential causal association between the elevated Lp(a) levels and an increased risk of ASCVD as well as calcific aortic valve stenosis (AS)10. Lp(a) is synthesised by the hepatocytes with core composition consisting of triglycerides, and cholesteryl esters surrounded by the outer membrane of phospholipids and free cholesterol. So, what makes Lp(a) so special? Lp(a) contains kringle IV (KIV) domains, followed by a single kringle V domain. These domains are consists of 10 different type of amino acid sequences9. Among these, KIV10 contains the site to which oxidised phospholipid attaches and KIV2 which may affect the concentration of Lp(a)11. Lp(a) is believed to promote foam cell formation, in addition to cause smooth muscle cell proliferation within the atherosclerotic plaque, causing turbulent blood flow, thereby destabilising the atherosclerotic plaque structure9 (Figure 1).
 in Youth - Endotext - NCBI Bookshelf-5.jpg)
Figure 1. Structure of lipoprotein(a) with repeating KIV domains linked through a S-S bond to apoB100. TG= triglycerides, CE= cholesterol ester, FC= free cholesterol, PL= phospholipid12
Studies have shown Lp(a) concentrations above 50 mg/dL being associated with coronary arterial disease (CAD)10. But has this been corroborated by larger studies? The answer to this is yes. In fact, Copenhagen City Heart Study performed on general population reported individuals with concentration of Lp(a) between 30 mg/dL and 76 mg/dL (67th-90th percentile) were at 1.6-fold increased risk of having myocardial infarction (MI) compared to individuals with Lp(a) levels below 5mg/dL. Furthermore, those with Lp(a) between 77 mg/dL to 117 mg/dL (90th to 95th percentile) were 1.90 likely to have an MI13. Lp(a) levels >180 mg/dL were correlated with risk of CV events similar to that seen in familial hypercholesterolemia14. Currently, there are no approved pharmacologic interventions that specifically target Lp(a) concentration, however, some therapies that primarily targets the LDL-C have also shown to affect the levels of Lp(a)13. Since there is no available treatment, what about screening everyone to determine their risks? The answer to this question is simple, it is not a viable option to screen everyone. In fact, the European Atherosclerosis Society, and the U.S. National Lipid Association recommended screening Lp(a) levels in those who are at intermediate to high risk of CVD. Notably, a once-in-a-lifetime Lp(a) measurement suffices with possible confirmatory repeated measurements in those with very high concentration15.
Considering ASCVD being the most prevalent cause of coronary heart disease (CHD), and the primary cause of mortality globally, LDL-C reduction is often a primary goal. Nonetheless, recently randomised controlled trials and real-world studies have shown that there are still residual risk of CV events even after LDL-C reduction16. Percutaneous coronary intervention (PCI) for instance is often necessary for CHD diagnosis and treatment, in addition to improve patient prognosis. In fact, the prevalence of PCI has increased from 34.1% in 1999 to 59.4% in 200717. Moreover, a high rate of recurrence adverse CV events, including MI, angina, heart failure, and sudden cardiac death after a PCI have been documented in the literature. Emerging studies have shown Lp(a) levels in patients with CHD after a PCI positively correlated with the occurrence of major adverse cardiovascular event (MACE) within 1 year16. In addition, a high Lp(a) levels is postulated to be the residual risk factor for MACE in individuals with LDL-C levels under 100 mg/dL16. Therefore, assessing Lp(a) level in these population groups post PCI may stratify their risk of recurrent CV events. Then what about those patients who have undergone coronary artery bypass grafting (CABG), does the risk persist?
In a study performed by Ezhov et al., 2011 where patients with chronic IHD (n=361) who underwent Lp(a) and lipid assessment prior to the CABG. These patients were then followed up for a mean of 66 +/- 34 months. In 109 of them, approximately 142 serious events including cardiac death (n=20), non-fatal MI (n=14), myocardial revascularisation (n=35), repeat CABG (n=6), and stroke (n=4) was reported. Interestingly, subjects with Lp(a) >30 mg/dL, the survival rate after CABG was lower (p<0.001). The study concluded in patients with chronic IHD, a high Lp(a) level may serve as an independent predictor of unfavourable events, including death and non-fatal MI 10 years after CABG18. These findings have also been substantiated by a study carried out by Yu et al., in 2023 where 5-year follow-up of patients on dual Ticagrelor plus aspirin antiplatelet strategy after coronary artery bypass grafting (DACAB) trial in which 500 patients who underwent primary isolated CABG were randomised to three-antiplatelet therapies for a year after surgery. Baseline Lp(a), and LDL-C levels were taken, and their levels were repeated during the follow-up (median follow-up time was 5.2 year)19. The results showed even though 28.1% patients achieved the desired LDL-C reduction (<1.8 mmol/L) during the follow-up period, there was still approximately 31.4% of cohorts with persistently elevated LDL-C levels (≥ 2.6 mmol/L). Furthermore, the postoperative MACE risk remained high in patients with a baseline Lp(a) ≥ 30 mg/dL, as was all-cause death (Figure 2). These results further substantiate the findings that a high baseline Lp(a) levels with synergistic effects of a high LDL-C is associated with a poor clinical outcome in patients who have undergone CABG19.
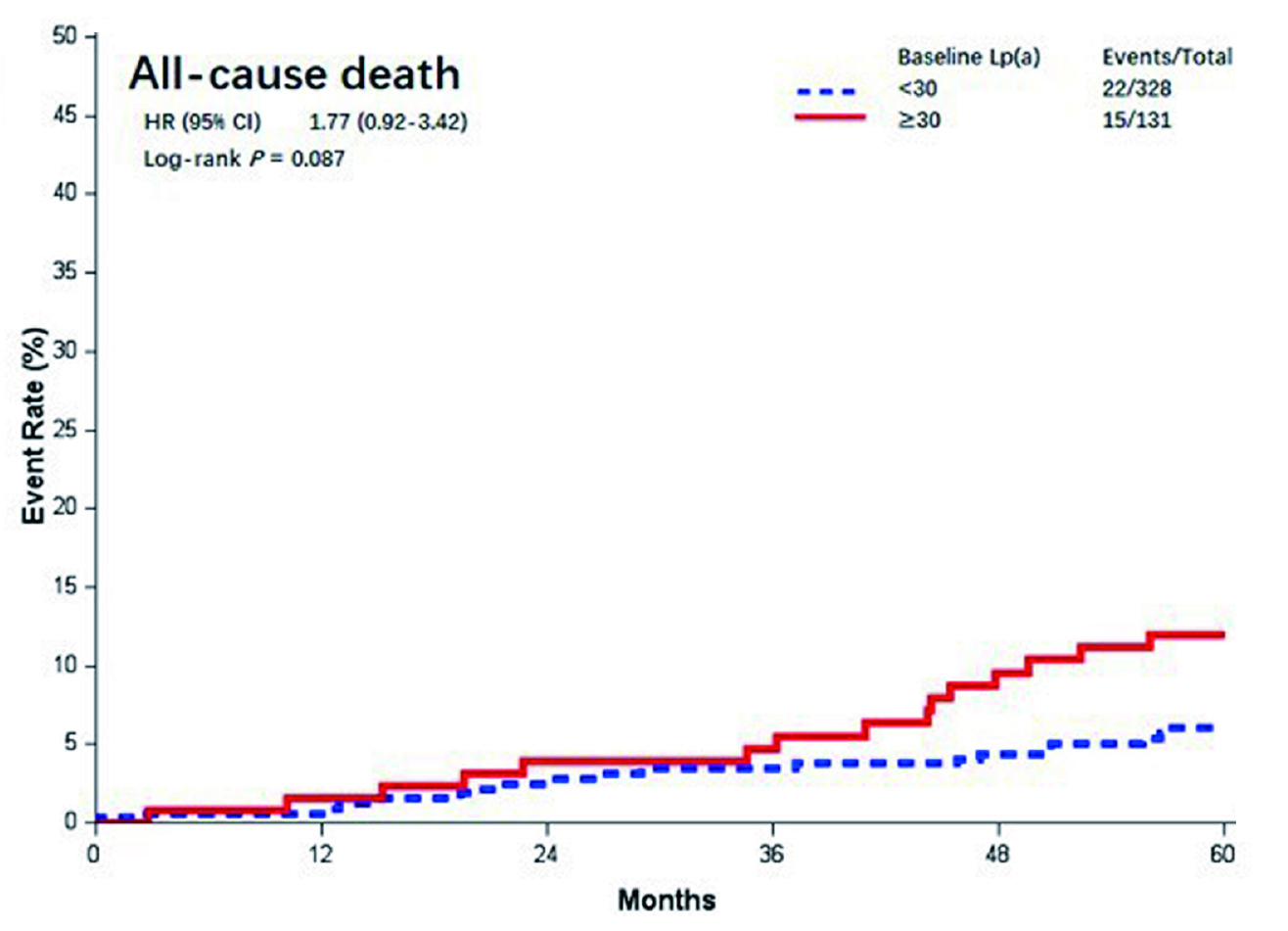
Figure 2. Kaplan-Meier curves of different baseline Lp(a) levels (mmol/l). Lp(a)= lipoprotein (a)19
Considering there are mounting evidence that Lp(a) is associated with CVD risk prediction, an appropriate therapeutic target is urgently needed. The most recent 2019 guidelines of the European Society of Cardiology and European Atherosclerosis Society (ESC/EAS) for management of dyslipidaemia recommended that Lp(a) measurement should be considered at least once to identify individuals with extremely elevated Lp(a) levels (>180 mg/dL or 430 nmol/L). Moreover, levels of Lp(a) appears to contribute to the stratification of patients with CVD risk between the moderate to high categories6. There are a number of potential Lp(a) lowering agents which have been studied with lipid apheresis and the use of antisense oligonucleotides being the most effective treatment modality in reducing Lp(a) levels up to 75% to 77.8%, respectively6.
More recent clinical studies such as ODYSSEY OUTCOMES trial showed 50% of acute coronary syndrome (ACS) patients having an Lp(a) level >21.2 mg/dL (~53 nmol/L) and post-hoc analysis also showed proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor is associated with significant Lp(a) reduction and CVD events by more than 23%20. Other potential agents include small interfering ribonucleic acid (siRNA) which specifically targets the Lp(a) messenger ribonucleic acid (mRNA) has shown reduce Lp(a) levels up to 90% without any severe adverse events in phase I trial. The phase 2 trial is still ongoing with results eagerly being awaited21. What about lifestyle changes, do they help? Well, according to recent studies, commonly used preventative strategies remains ineffective against Lp(a) reduction, most notably, the lifestyle changes including diet and exercise do not significantly affect Lp(a) levels since 75% to 95% of cases with elevated Lp(a) levels are predominately inherited, affected by single Lp(a) gene22,23. In conclusion, Lp(a) represents an exciting new biomarker, and routine testing in currently not advocated. Nonetheless, testing those at high risk of CVD may help improve CV outcomes among these individuals.
References
1. Kim H, Kim S, Han S, et al. BMC Public Health 2019; 19(1): 1112. 2. Pahwa R, et al. StatPearls Publishing LLC.; 2023. 3. Jung E, et al. Int J Environ Res Public Health 2022; 19(14). 4. Rong S, et at. J Am Heart Assoc 2022; 11(15): e023690. 5. Zhu L, et al. BMC Cardiovascular Disorders 2022; 22(1): 474. 6. Parthymos I, et al. European Journal of Preventive Cardiology 2021; 29(5): 739-55. 7. Farzam K, et al. StatPearls Publishing LLC.; 2023. 8. Zafrir B, et al. Int J Cardiol Cardiovasc Risk Prev 2023; 16: 200173. 9. Rhainds D, et al. Current Atherosclerosis Reports 2021; 23(9): 51. 10. Duarte Lau F, et al. JAMA Cardiology 2022; 7(7): 760-9. 11. Coassin S, et al. Eur Heart J 2017; 38(23): 1823-31. 12. McNeal CJ, et al. MDText.com, Inc 2000. 13. Kronenberg F. Springer International Publishing; 2022: 201-32. 14. Ugovšek S, et al. Biomolecules 2021; 12(1). 15. Kamstrup PR. Clinical Chemistry 2020; 67(1): 154-66. 16. Zhang J, et al. Am J Transl Res 2023; 15(1): 165-74. 17. Mokadam NA, et al. J Card Surg 2011; 26(1): 1-8. 18. Ezhov MV, et al. Kardiologiia 2011; 51(1): 18-22. 19. Yu Q, et al. Front Cardiovasc Med 2023; 10: 1103681. 20. Szarek M, et al. Eur Heart J 2020; 41(44): 4245-55. 21. Nurmohamed NS, et al. Curr Atheroscler Rep 2022; 24(11): 831-8. 22. Enkhmaa B, et al. Nutrients 2020; 12(7). 23. Trinder M, et al. JAMA Cardiology 2021; 6(3): 287-95.