Oncolytic Herpes Simplex Virus An Emerging Solution for Glioblastoma
Glioblastoma (GBM) is one of the most aggressive and deadliest cancers with only few patients survive 2.5 years and less than 5% of patients survive 5 years following diagnosis1. Unfortunately, the treatment outcomes of current treatment approaches are modest and effective solutions for the disease are desirable. There is growing evidence supporting the safety and oncolytic activity of unarmed and armed oncolytic herpes simplex virus (oHSV) combined with existing therapies. With persistent endeavour to advance the engineering of oHSV, the armed oncolytic virus would be a potential cure of this fatal disease.
Fighting GBM - an Ongoing Challenge
Glioma accounts for 81% of malignant brain tumours while GBM is the most common type of glioma characterised by its highly infiltrative, angiogenic, heterogenous and immunosuppressive nature2. Current treatment approaches entailing supramarginal resection followed by adjuvant radiotherapy and temozolomide (TMZ) chemotherapy yield suboptimal outcomes that median overall survival (OS) of GBM remains 12-15 months3. The universal local recurrence, resistance to TMZ and ionising radiation conferred by the expression of O6-methytlguanine-DNA-methyltransferase4 and glioblastoma stem-like cells (GSCs)5 are formidable challenges that highlight a pressing need for new treatment strategies.
oHSV has been extensively researched as a potential cure of GBM. Given its naturally neurotropic and episomal nature with superior oncolytic activity to adenovirus6 and effective anti-viral drugs available7, oHSV is considered an advantageous candidate of virotherapy for GBM. By replicating selectively in tumour cells whose antiviral response via the double-strand RNA-dependent protein kinase-eukaryotic initiation factor 2α pathway is defective, infected tumour cells are lysed by virus-mediated cytotoxicity, hence innate and adaptive immune systems are awakened (Figure 1)8.
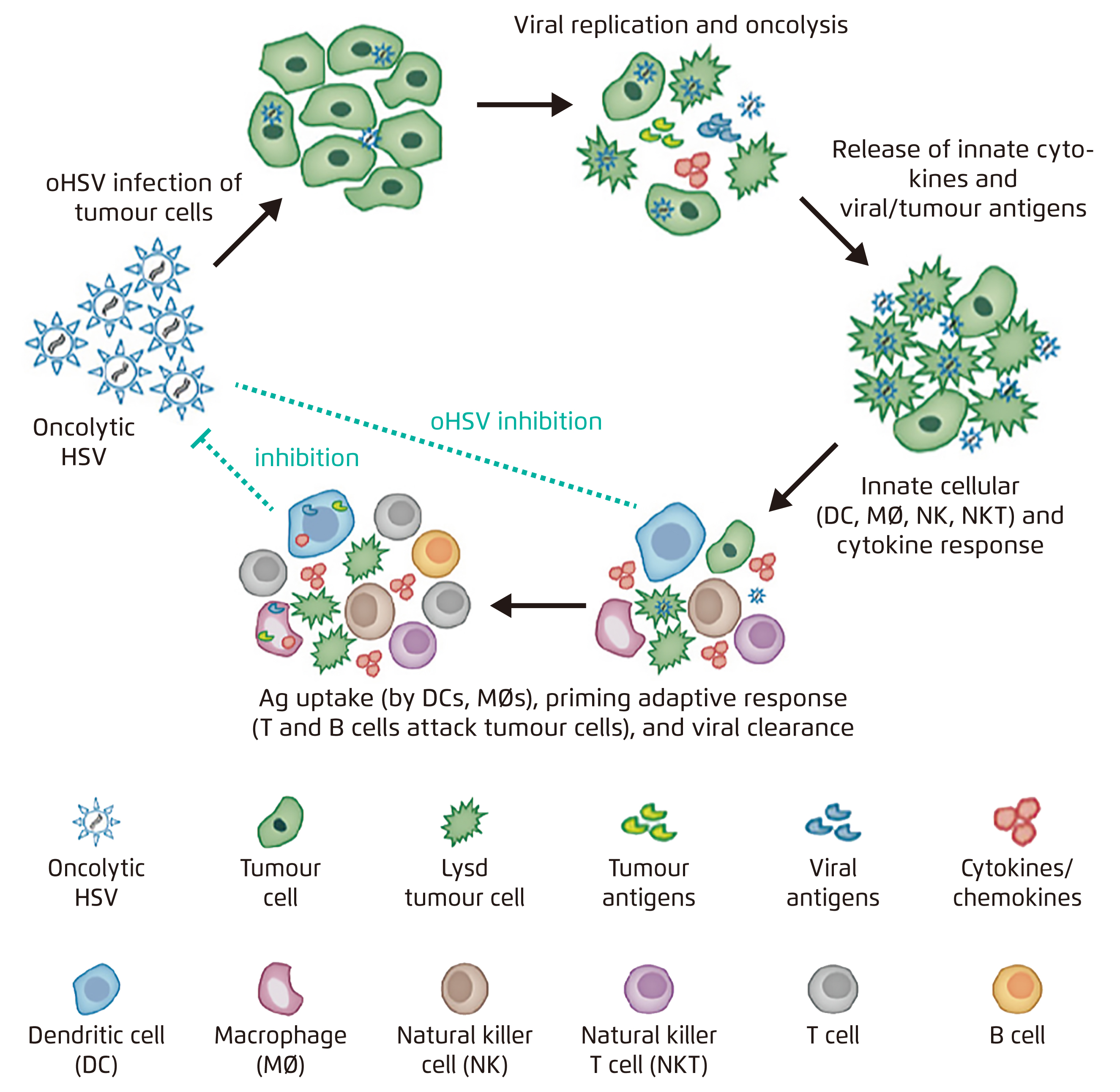
Figure 1. Mechanism of action of oHSV8
Evolution of Unarmed oHSV
The tactics of genetic manipulations to attenuate neuropathogenicity and enhance replication selectivity and antitumoral immune responses have been evolving over the past decades, giving rise to multiple unarmed oHSV. The preclinical development of unarmed oHSV is outlined in Figure 2.

Figure 2. Timeline of preclinical development of unarmed oHSV
The mutant HSV1716 was developed in 1991, it has a 759-bp deletion of γ34.5 to eliminate neurovirulence9. Further in 1995, the mutant G207 is designed by deleting both γ34.5 loci and inserting a lacZ marker gene in the viral infected cell protein 6 (ICP6) gene, inactivating viral ribonucleotide reductase. The multiple mutations of HSV-1 G207 minimise the chance of reversion to wild-type virus and confer additional safety features such as sensitivity to ganciclovir and temperature sensitivity, which halts viral activity in the febrile host10.
Engineered from G207, G47∆ has an additional deletion within α47 gene which downregulates major histocompatibility complex class I expression on infected cells. This deletion also counteracts γ34.5 deletion, accelerating viral life cycle and producing higher titers of HSV progeny. The potency and anti-tumour activity of G47∆ were demonstrated to be significantly improved in animal models as compared with G20711. Further, it also infects and kills GSCs in hypoxic tumour microenvironment and inhibits their self-renewal capacity, reducing the chance of recurrence12. Besides as monotherapy, G47∆ was reported to show synergistic efficacies against GSCs in combined with TMZ13 or etoposide14.
Although the safety has been proven in clinical trials, γ34.5-null viruses grow poorly in cells15. Hence, the glioma-selective HSV-1 mutant, rQNestin34.5, was engineered by expressing γ34.5 under control of a synthetic nestin promoter, whereas expression of the intermediate filament nestin is a molecular marker of malignant gliomas. Essentially, rQNestin34.5 was shown to double the survival of mice bearing glioma15.
From HSV1716 to rQNestin34.5, from the manipulation of a single gene (HSV1716) to multiple genes (G207, G47∆, rQNestin34.5), there has been a tremendous effort to enhance replicative capacity and selectivity without compromising the safety of unarmed oHSV. While preclinical studies demonstrated improved outcomes as the strategies of genetic modification advance, results from the ongoing clinical trials are anticipated.
Arming oHSV to Achieve Anti-Tumour Effects
Arming oHSV to express different therapeutic genes opens up many possibilities and is a promising approach to deliver various anti-tumour effects. For instance, G47∆-mIL12 is armed with murine interleukin (IL) 12 which triggers ischaemic necrosis of tumour16. G47∆-mIL12 shows versatile effects in a GBM model including direct oncolysis, depletion of local regulatory T cells (Tregs), stimulation of T-helper 1 (Th1) immunity, inhibition of tumour angiogenesis, killing of GSCs, stimulation of interferon γ production and downregulation of vascular endothelial growth factor16. Besides, the triple combination therapy with systemic anti-programmed cell death-1 (PD-1) and anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) antibodies further improves effector T cells to Tregs ratio and increases M1-like macrophage polarisation compared to G47∆-mIL12 alone, resulting in 89% of treated mice surviving long term and protected by immunological memory (Figure 3)17.
Apart from anti-angiogenic armed oHSV, G47∆-TRAIL expressing secretable tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is an example of pro-apoptotic armed oHSV. It inactivated mitogen-activated protein kinase (MAPK) signaling, upregulates c-Jun N-terminal kinase and p38-MAPK signaling and induces cleaved caspase 3, promoting apoptosis even in resistant GBM cells. It prolonged the survival of mice with intracranial tumours derived from TMZ-resistant primary and recurrent GSC18.
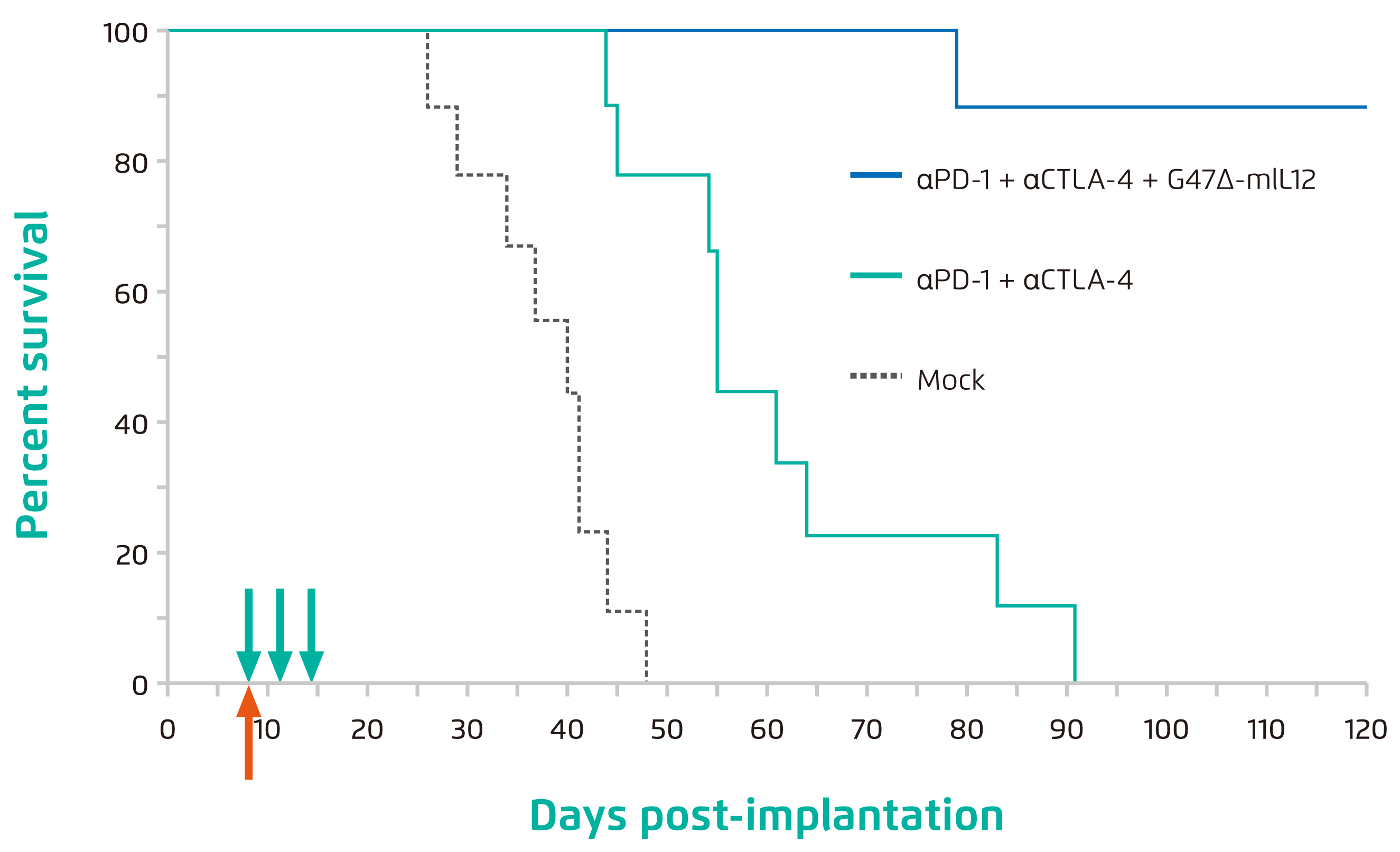
Figure 3. Percent survival of mice post implantation of 005 GSC-derived GBM17,
Mock: mice treated with isotype control antibodies
Future Perspectives
Despite the effort to improve the replication capacity of oHSV, premature viral clearance is a frequent obstacle as a result of host innate immune response primarily mediated by the natural killer (NK) cells, microglia and macrophages19. Hence, further optimisation on transient immunosuppression and administration protocol of virotherapy involving oHSV is needed. For instance, investigations on the route of delivery for achieving the optimal tumour-specific delivery and intratumoural spread while protecting the virus from immunosurveillance is crucial. Of importance, the genetic profile of GBM needs to be better characterised to identify molecular targets for developing retargeted oHSV and precision medicine.
Acknowledgement
The editorial support by Ms Anna Yau of the School of Biomedical Sciences, CUHK in preparing this article is deeply appreciated.
Reference
1. Tamimi et al. Glioblastoma. Codon Publications; 2017:143-153. 2. Saha et al. Immunotherapy. 2018. 3. van Linde et al. J Neurooncol. 2017;135(1):183-192. 4. Hegi et al. N Engl J Med. 2005;352(10). 5. Bao et al. Nature. 2006;444( 7120):756-760. 6. Hoffmann et al. Cancer Gene Ther. 2007;14(7). 7. IWAMI et al. Neurol Med Chir (Tokyo). 2010;50(9):727-736. 8. Bommareddy et al. Annu Rev Cancer Biol. 2018;2(1):155-173. 9. Randazzo et al. Virology. 1995;211(1):94-101. 10. Cinatl et al. Neoplasia. 2004;6(6):725-735. 11. Todo et al. Proc Natl Acad Sci. 2001;98(11):6396-6401. 12. Wakimoto et al. Cancer Res. 2009;69(8):3472-3481. 13. Kanai et al. J Virol. 2012;86(8):4420-4431. 14. Cheema et al. Clin Cancer Res. 2011;17(23):7383-7393. 15. Kambara et al. Cancer Res. 2005;65(7):2832-2839. 16. Cheema et al. Proc Natl Acad Sci. 2013;110(29):12006-12011. 17. Saha et al. Cancer Cell. 2017. 18. Tamura et al. Mol Ther. 2013;21(1):68-77. 19. Alvarez-Breckenridge et al. Nature 2012. 20. Fulci et al. Cancer Res. 2007;67(19).