
Specialist in Haematology
AL Amyloidosis: A Rare but Multifaceted Disease
Light chain (AL) amyloidosis is a rare multi-systemic, incurable protein misfolding disorder characterised by plasma cell dyscrasia and abnormal clonal plasma cell proliferation1. Immunoglobulin secreting cells typically produce a monoclonal light chain of kappa (κ) or lambda (λ) type, and the protein normally conforms to an alpha-helical configuration2. Nonetheless, when light chain protein misfolds, they form the beta-pleated sheets which can deposit in tissues and interfere with organ function, causing multi-organ dysfunction1,2. Notably, 70% of patients with AL amyloidosis have cardiac involvement and 68% have multiple organ involvement at the time diagnosis3. When detected at a late stage, AL amyloidosis carries a poor long-term prognosis with a median survival rate as short as 5 months with infection, cardiac or hepatic failure being the most common cause of death4.
Of note, majority of patients with AL amyloidosis are elderly, with a mean age at diagnosis being 63 years1. Moreover, AL amyloidosis is relatively rare with an incidence rate of 14.3 cases per million population (PMP) reported in Japan, 10.01 and 9.97 cases PMP reported in Taiwan and South Korea, respectively1. Due to the insidious nature of the disease, the clinical manifestations are non-specific, hence, the diagnosis often occurs at later stages of the disease after critical organ damage rendering anti-amyloid treatment ineffective1. Therefore, to understand the burden of AL amyloidosis and the current unmet needs, we have invited Dr. Raymond Wong, haematology specialist to discuss the impact of the disease on patients, in addition to share his expertise in treating AL amyloidosis.
Understanding the Enigma of AL Amyloidosis
Similar to multiple myeloma (MM), AL amyloidosis typically affects males more than females (3:2 ratio) with no ethnic or geographic specificity4. The local prevalence of the disease is not well documented since AL amyloidosis has traditionally been seen as a rare disease, and there is no specific registry available on AL amyloidosis locally, Dr. Wong explained. Of note, AL amyloidosis is considered as the most severe form of systemic amyloidosis and the amyloid deposits containing the light chains infiltrate tissues and causes organ dysfunction and failure5. Although amyloids can effect any organs, it has a predilections for specific organs which include kidneys in 74% of cases that can lead to the development of nephrotic syndrome, followed by renal failure in some cases5. Other organs commonly affected by the amyloids include the heart (60%), gastrointestinal tract (10-20%), liver (27%) and autonomic nervous systemic (18%)5. Importantly, cardiac involvement is considered as the main prognostic determinant in AL amyloidosis and is the leading cause of death, particularly in those diagnosed late and those not responding to the treatment5.
The present medical challenges is primarily related to the fact that the symptoms of AL amyloidosis may mimics those of the other conditions, leading to a delay in diagnosis and ensuing unmet clinical needs for the affected individual6. In relation to this, Dr. Wong elaborated that the median time from symptoms onset to correct diagnosis has been reported to take up 2 years7 and patients are often diagnosed after their disease has progressed to a more advanced stage with organ damage6. In comparison, the median survival rate of patients with very advanced cardiac disease secondary to AL amyloidosis is only 4 months compared to about 2 years among patients with less severe cardiac involvement7. In fact, studies have shown that 44% of patients are initially misdiagnosed and treated as having unrelated cardiomyopathy, with 3 out of 4 cardiologist reporting misdiagnosis due to a poor disease awareness7. Regarding this, Dr. Wong added that patients with AL amyloidosis are at increased risk of developing heart failure and cardiac arrythmias8, but since these symptoms are not specifically related to AL amyloidosis7, the diagnosis of AL amyloidosis is only evident after further investigations9. To diagnose amyloidosis, biopsy of a clinically affected organ is often the most sensitive method, and may also help detect other pathologies5.
However, kidney or heart biopsies, are invasive and associated with an increased risk of bleeding5. Therefore, if amyloidosis is clinically suspected, a less invasive method is preferred, for instance, serum as well as urine protein electrophoresis with immunofixation, serum free light chain assays and bone marrow sampling can be performed which could help to support the diagnosis10. The gold standard of AL diagnosis is through tissue typing using mass spectrometry but this method is not currently available locally, instead traditional histopathological diagnosis of amyloidosis based on Congo red dye to amyloid is used to identify the amyloid fibrils11, according to Dr. Wong.
Burden of AL Amyloidosis and the Current Treatment Unmet Needs
The precise aetiology of AL amyloidosis is poorly understood, but some studies have suggested that a high circulating levels of monocytes and tumour necrosis factor receptor superfamily member 17 (TNFRSF17) protein being the risk factors for the disease12. On a more important note, patients with AL amyloidosis often experience substantial disease burden that can lead to impairment in daily functioning. Symptoms and complications depend on the number and types of organ systems involved and the duration of time between symptom onset and treatment3. Moreover, AL amyloidosis can lead to anxiety, frustration and depression as patients are often left to grapple with the gravity and rarity of their condition3. Therefore, Dr. Wong emphasised that it is important to offer the appropriate treatment and lifestyle modification which can help improve the patient’s well-being, overall health-related quality of life3 and maximise the functional status13.
Notably, the survival rate of patients with AL amyloidosis is poor since the organ damage is often irreversible and mortality is primarily driven by cardiac failure14. Hence, the primary goal of the treatment is to recover the organ function by targeting the abnormal plasma cell clones promptly using treatment regimens from those used to treat MM14. Regarding the traditional treatment of AL amyloidosis, Dr. Wong elaborated that AL amyloidosis is an uncommon disorder and given patients’ frailty and the high early mortality rate, the management remains challenging15.
This is further potentiated by the lack of approved therapies for AL amyloidosis16 and in the past, patients with AL amyloidosis were treated with therapies that were borrowed and customised from the treatment of MM15; for instance, the combination of bortezomib, dexamethasone and cyclophosphamide (CyBorD) or melphalan (BMDex) have been used to treat patients with AL amyloidosis17. Even though CyBorD is an effective regimen for treating patients with newly diagnosed AL amyloidosis, it remains inadequate to enhance the outcomes in high-risk groups18. Furthermore, patients with AL amyloidosis treated with these agents often experience more frequent and severe toxicity compared to patients with MM receiving the same regiments. Therefore, the unmet need remains for a more tolerable and effective therapy for AL amyloidosis19.
An Evolution in AL Amyloidosis Treatment
Although autologous stem cell transplantation (ASCT) is a treatment option for AL amyloidosis, but having more than two organs involved has been considered a high risk factor for ASCT, particularly in those with cardiac involvement20. Very recently, daratumumab, which is a human IgG-κ monoclonal antibody that targets CD38 expressed on the human plasma cells in MM has shown some promising results in patients newly diagnosed with AL amyloidosis21. Regarding daratumumab use in AL amyloidosis, Dr. Wong commented that daratumumab has an excellent safety and efficacy, in addition to being well tolerated by patients newly diagnosed with AL amyloidosis22.
Furthermore, patients treated with daratumumab achieved rapid haematological response that deepened over time with no unexpected side-effects from neither daratumumab itself nor from any of the combination partners that are used23. The findings reported by Dr. Wong were also substantiated in the ANDROMEDA trial, which included 388 patients newly diagnosed with AL amyloidosis21. They were randomised to receive six cycles of bortezomib, cyclophosphamide, and dexamethasone either alone (control group) or with subcutaneous daratumumab, followed by a single agent daratumumab every 4 weeks for up to 24 cycles (daratumumab group) with a median follow-up period of 11.4 months21. Haematologic complete response was the primary endpoint and the percentage of patients achieving the primary endpoint was significantly higher in the daratumumab group compared to the control group (53.3% vs 18.1%) (relative risk ratio of 2.9; 95%, confidence interval [Cl]: 2.1-4.1; p<0.001)21. Moreover, the survival free from major organ deterioration or haematologic progression favoured the daratumumab group (hazard ratio of 0.58; 95%, CI: 0.36-0.93; p= 0.02) (Figure 1)21.
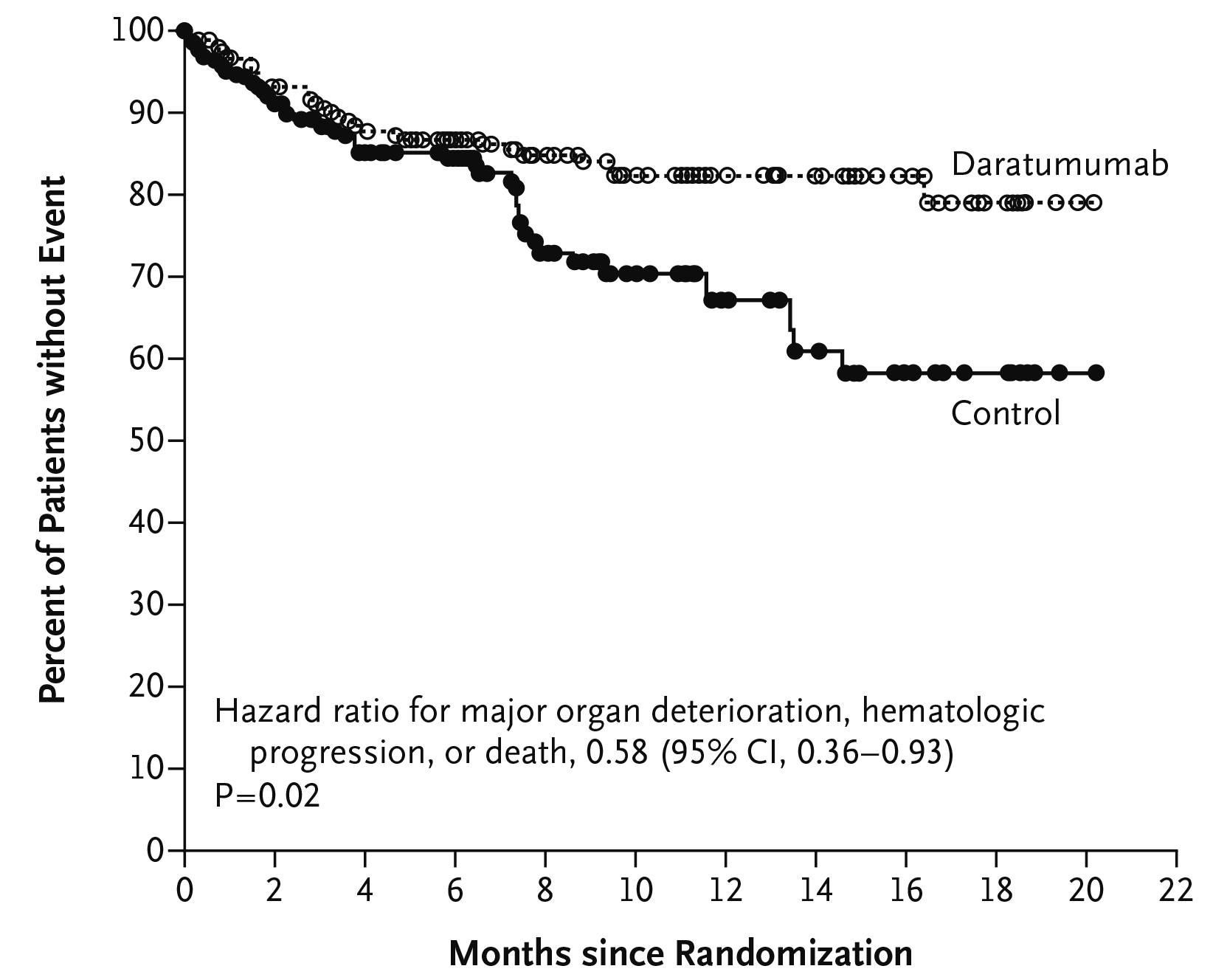
Figure 1. Kaplan-Meier Estimates of Survival Free from Major Organ Deterioration or Haematologic Progression21. CI= confidence interval.
Remarkably, at 6 months, more cardiac and renal responses occurred in the daratumumab group compared to the control group (41.5% vs 22.2% and 53.0% vs 23.9%, respectively)21. The study concluded that among patients with newly diagnosed AL amyloidosis, addition of daratumumab to CyBorD was associated with a higher frequency of haematologic complete response, and survival free from major organ deterioration or haematologic progression21. Importantly, in the past, there was a treatment gap in AL amyloidosis since there was a deficient in a therapeutic agent which was well-tolerated, effective, safe, and capable of reducing free light chains (FLCs) rapidly23. However, these unmet needs have been fulfilled by the addition of daratumumab, which is the one and only approved treatment for patients newly diagnosed with AL amyloidosis23. Therefore, Dr. Wong advocated not to withhold such treatment from those with features of AL amyloidosis and those with pre-existing organ dysfunction secondary to AL amyloidosis since the disease can have a detrimental effect on their long-term outcome.
References
1. Kumar N, et al. Orphanet J Rare Dis 2022; 17(1): 278. 2. Gertz MA. American Journal of Hematology 2022; 97(6): 818-29. 3. Bayliss M, et al. Orphanet J Rare Dis 2017; 12(1): 15. 4. Baker KR. et al. Methodist Debakey Cardiovasc J 2022; 18(2): 27-35. 5. Ryšavá R. Nephrology Dialysis Transplantation 2018; 34(9): 1460-6. 6. Sabinot A, et al. Blood Reviews 2023; 59: 101040. 7. Wechalekar AD, et al. JACC: CardioOncology 2022; 4(4): 427-41. 8. Wechalekar AD, et al. JACC: CardioOncology 2022; 4(4): 427-41. 9. De Michieli L, et al. Intern Emerg Med 2023; 18(7): 1879-86. 10. Melmed GM. et al. Proc (Bayl Univ Med Cent) 2009; 22(3): 280-3. 11. Muchtar E, et al. Acta Haematologica 2016; 135(3): 172-90. 12. Saunders CN, et al. Blood Adv 2021; 5(13): 2725-31. 13. Hassan H, et al. Hemato 2022; 3(1): 38-46. 14. Quock TP, et al. J Comp Eff Res 2023; 12(2): e220185. 15. Muchtar E, et al. Mayo Clin Proc 2021; 96(6): 1546-77. 16. Lousada I. Orphanet J Rare Dis 2020; 15(1): 268. 17. Kastritis E, et al. Blood Adv 2019; 3(20): 3002-9. 18. Diaz-Pallares C, et al. Clin Lymphoma Myeloma Leuk 2020; 20(6): 394-9. 19. Palladini G, et al. Blood 2020; 136(1): 71-80. 20. Al Saleh AS, et al. Biol Blood Marrow Transplant 2019; 25(8): 1520-5. 21. Kastritis E, et al. New England Journal of Medicine 2021; 385(1): 46-58. 22. Ehsan H, et al. Clinical Lymphoma Myeloma and Leukemia 2022; 22(5): e285-e92. 23. Jeryczynski G, et al. ESMO Open 2021; 6(2): 100065.